Amyotrophic lateral sclerosis (ALS) represents the kiss of death, striking its unsuspecting victims without warning. It affects about 4.4 people per 100,000; they gradually lose the ability to control their muscles, until eventually even breathing becomes impossible. ALS is the most common of a collection of neurodegenerative diseases that affect the motor neurons, the cells that control the movement of muscles. It is always terminal, with an average survival time of just two to four years.
Even the most extraordinary of individuals, such as the British theoretical physicist Stephen Hawking, could not elude its clutches. His exceptional intelligence found itself imprisoned within a body ravaged by ALS, ultimately able to communicate only through a speech-generating apparatus he manipulated by twitching his cheek. Researchers have long looked into the intricate mechanisms of this devastating disease, endeavouring to unravel the mysteries behind it.
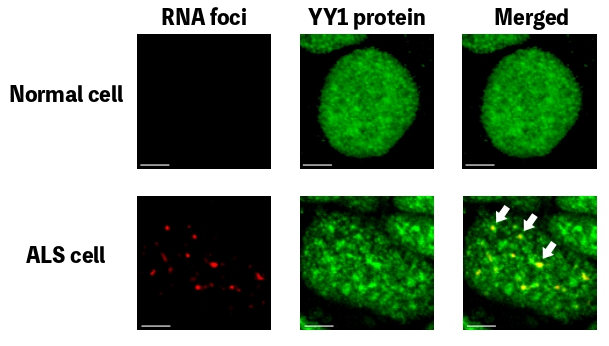
At the moment, the disease is uncurable, but Professor Edwin Chan Ho-yin and his research team from the School of Life Sciences at The Chinese University of Hong Kong (CUHK) are trying to do something about that. To be able to develop drugs that can potentially treat the disease, first we need to know more about what causes it – and that’s what the team has been searching for. In research that has been published in leading journal Nature Communications, they single out a trouble maker: Yin Yang 1 (YY1), a transcription factor, which is a type of protein that controls the transcription of genetic information to messenger RNA.
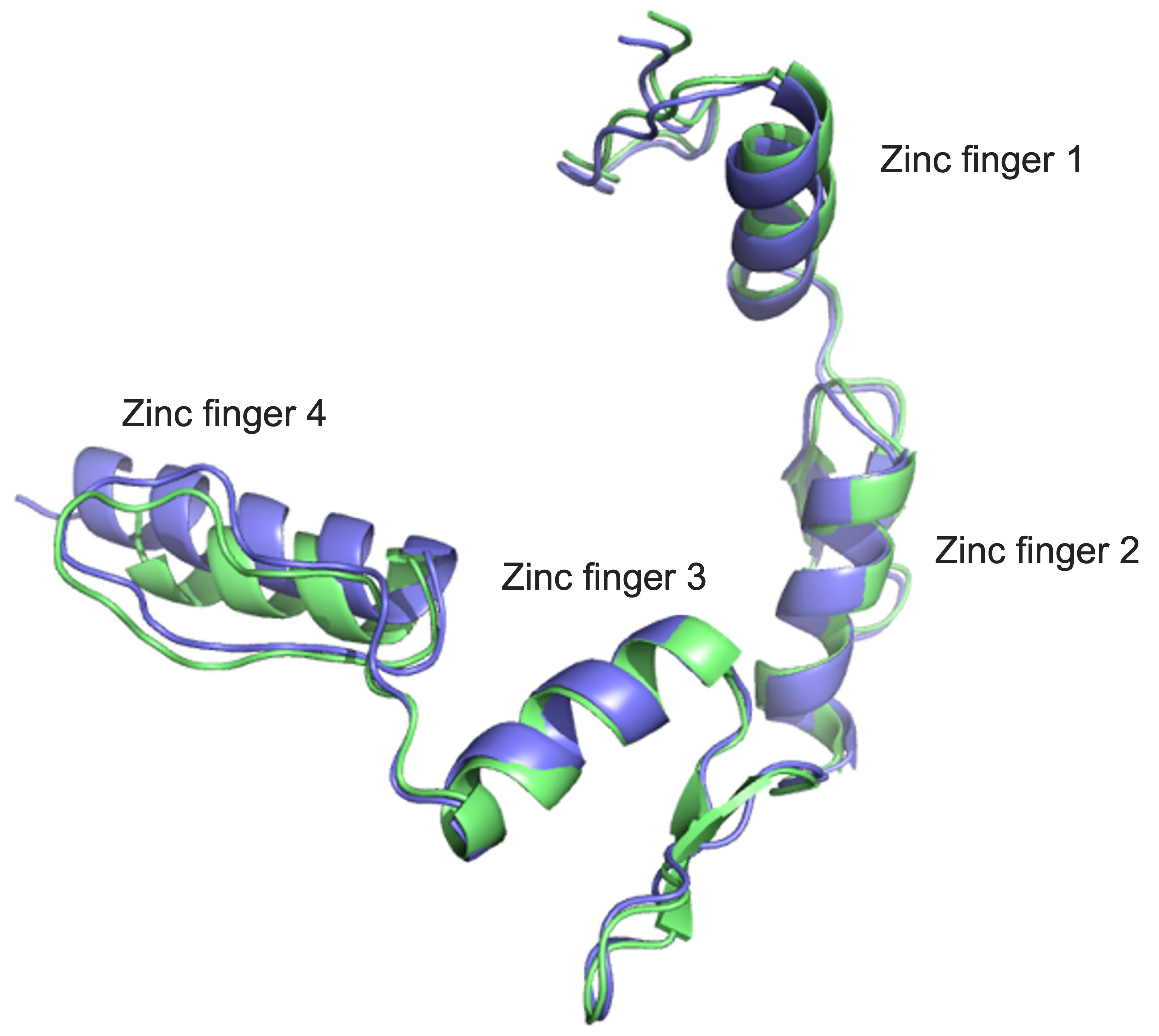
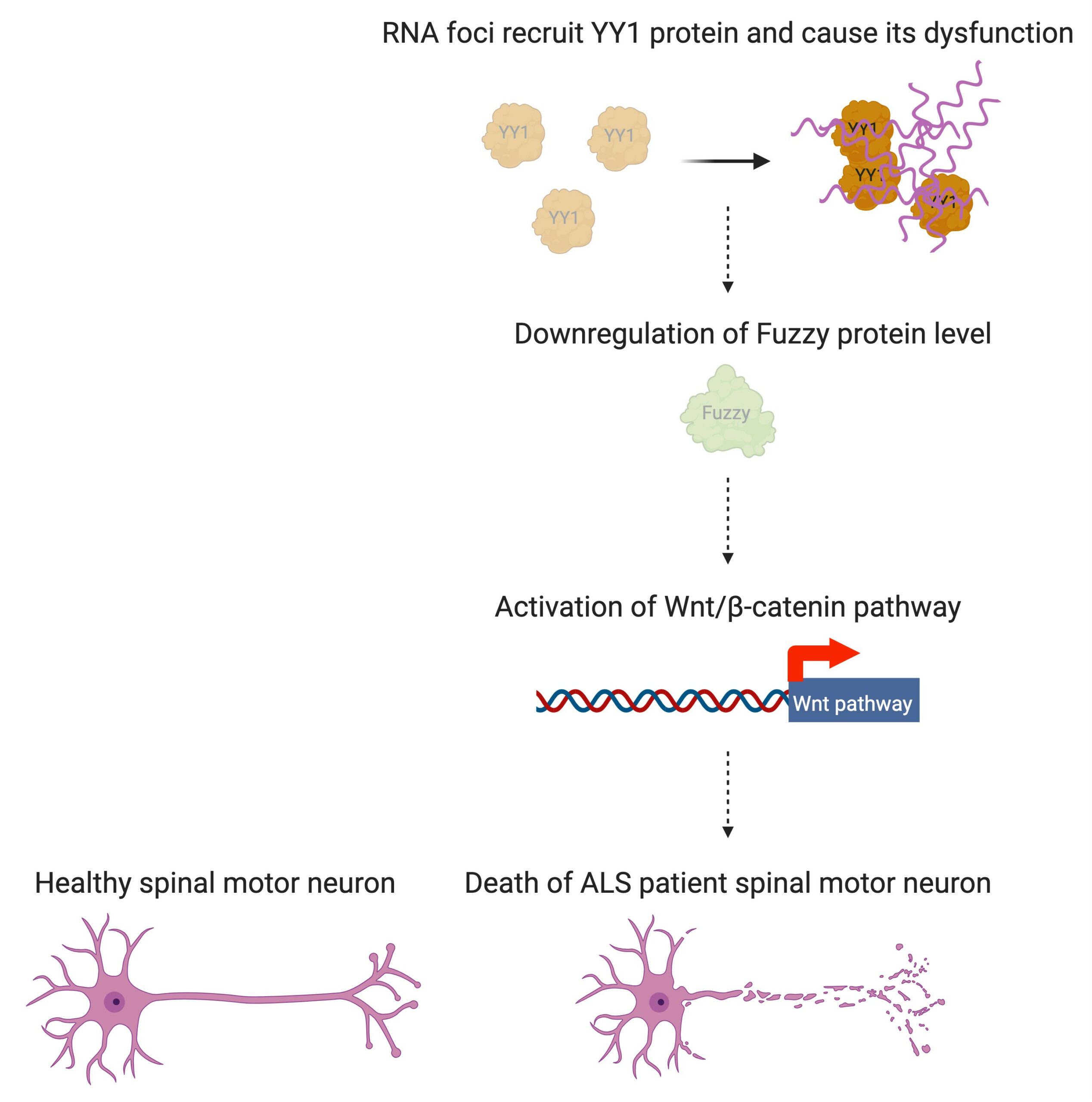
In people with ALS, for genetic reasons, the research team discovered, YY1 just doesn’t work so well in their spinal motor neurons, with RNA foci forming within them. “By perturbing the RNA/YY1 association, the YY1 protein is not able to carry out its normal function in neurons, causing neurodegeneration,” says Professor Chan. It hampers the expression of a gene known as Fuzzy, which YY1 is in charge of transcribing, which in turn causes neuronal cells to die. Inhibiting this pathway has a positive impact on the dysfunctional motor neurons.
The research is a collaboration with the University of Oxford, specifically Professor Kevin Talbot’s laboratory at Oxford’s Nuffield Department of Clinical Neurosciences. The partnership, which began in 2018, grew out of the Lee Hysan Postdoctoral Fellowship scheme in Clinical Neurosciences, with Stephen Chen, one of Professor Chan’s students, undertaking a fellowship in Professor Talbot’s laboratory.
To make the project happen, Chen set up induced pluripotent stem cell or iPSC-derived neuron reprogramming and differentiation technologies at CUHK after returning from Oxford. The move also stands it in good stead for the future, allowing it to perform routine patient neurons experiments on site.
After identifying one of the mechanisms that causes ALS, Professor Chan and his team started to create and screen molecules that might be able to treat ALS. They started working in 2018 on a prototype of the BIND family of peptide drugs, screening about 100 different versions of BIND before finally coming up with a 21-amino acid long peptide molecule that can potentially be used to treat the condition. It’s part of a family of medicines that have already proven useful in treating a range of other extremely unpleasant, incurable, inherited neurodegenerative conditions, including Huntington’s Disease and spinocerebellar ataxias. The version used in the current research, though, has been adapted for ALS – specifically, a subtype of the disease known as C9 ALS, which is one of some 34 subtypes of genetically caused ALS so far identified, but which is responsible for about 40% of cases.

“When we treated these neurons with our BIND peptide drug, which can stop the RNA foci from recruiting YY1, they restored their normal functioning, including the ability to make connections – synapses – with each other,” says Professor Chan. “Synapse formation can be used to measure how good the communication is between nerve cells in the brain. In ALS and other neurodegenerative diseases, synapse formation is perturbed, which results in neuronal defects and results in locomotor symptoms.”
So far, the team has tested drugs in cells and in fruit flies. Next up are tests on rodents and primates: the drug is ready to go, just requiring approval for the trials. They are also currently developing a second family of peptide drugs that can be used to treat ALS. Additionally, there are many other pathways that trigger the condition, which the team are also attempting to identify. If they can do so, they can potentially open the floodgates for other drugs to be developed that can help alleviate the life-threatening misery of the world’s ALS sufferers.